Article Text

Abstract
Introduction Evidence from randomised controlled trials on anti-tumour necrosis factor (TNF) agents in patients with Behçet’s syndrome (BS) is low.
Method We conducted a phase 3, multicentre, prospective, randomised, active-controlled, parallel-group study to evaluate the efficacy and safety of either infliximab (IFX) or adalimumab (ADA) in patients with BS. Adults patients with BS presenting with active mucocutaneous manifestations, occurring while on therapy with either azathioprine or cyclosporine for at least 3 months prior to study entry, were eligible. Participants were randomly assigned (1:1) to receive IFX or ADA for 6 months. The primary study outcome was the time to response of manifestations over 6-month anti-TNF alpha agents’ treatment.
Results 42 patients underwent screening visits, of whom 40 were randomly assigned to the IFX group (n=22) or to the ADA group (n=18). All patients at the time of randomisation had active mucocutaneous manifestations and a smaller proportion had concomitant vital organ involvement (ie, six and three patients with ocular and neurological involvement, respectively). A total of 14 (64%) responders in the IFX group and 17 (94%) in the ADA group were observed. Retention on treatment was 95% and 94% in the IFX and in the ADA group, respectively. Quality of life resulted to be significantly improved in both groups from baseline, as well as Behçet’s Disease Current Activity Form assessment. We registered two adverse events (one serious) in the ADA group and three non-serious adverse events in the IFX group.
Discussion The overall results of this study confirm the effectiveness of both IFX and ADA in achieving remission in patients with BS affected by mucocutaneous involvement.
- Infliximab
- Adalimumab
- Uveitis
- Behcet Syndrome
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Over the last two decades, the beneficial effects of anti-tumour necrosis factor (TNF) alpha agents, either infliximab (IFX) and adalimumab (ADA) have been amply demonstrated, not only for severe and refractory ocular disease, but also for treating almost all the systemic manifestations of Behçet’s syndrome (BS). The pharmacological management of patients with BS is based on the 2018 European Alliance of Associations for Rheumatology recommendations, which indicate that monoclonal anti-TNF antibodies should be considered in severe disease as first-line or in refractory patients. Additional data emerged from trials and large cohort studies were published after the 2018 recommendations, further confirming the efficacy and safety of ADA and IFX However, the conducted trials covered a single anti-TNF-alpha agent (either ADA or IFX and never together in the same study), and were focused on a specific organ involvement (eg, intestinal or ocular).
WHAT THIS STUDY ADDS
In this randomised clinical trial that involved 40 patients, the overall results confirmed the efficacy and safety of both ADA and IFX in achieving remission in patients affected by mucocutaneous involvement, confirming the effectiveness in the proportion of patients also with associated organ vital involvement due to BS, refractory to traditional immunosuppressive therapy. In this study, both ADA and IFX showed an excellent retention on treatment, and safety profile. In terms of efficacy, the occurrence of mucocutaneous manifestations, as well as the smaller proportion of ocular or neurological, during follow-up, that were defined as relapses, were seen in a minority of patients, both in the ADA and in the IFX group. Also, all the dimensions of quality of life as assessed by Short Form Health Survey 36 (SF-36) resulted to be significantly improved in both groups from baseline, as well as the assessment of disease performed through the Behçet’s Disease Current Activity Form.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Collecting further evidence on the effectiveness of both drugs in BS can be considered an important milestone also for encouraging the health authorities to speed up the procedures for approving their on-label use. Indeed, both drugs demonstrated robust improvement in clinical response, quality of life, besides a high level of adherence to treatment and tolerability. These results are therefore also crucial in terms of prescriptiveness (currently off-label), as the evidence coming from real life needed to be confirmed by clear data from clinical trials.
Background
Behçet syndrome (BS) is a rare, chronic and multisystemic disease, whose onset more frequently occurs in the late third and early fourth decades of life. It may present with recurrent oro-genital ulcers, ocular inflammation and skin manifestations.1 Joint, vascular, gastrointestinal and neurological involvement can also occur.2 The clinical picture of the disease is variable; while prevalent mucocutaneous involvement and arthritis represent the only clinical features in subjects with a benign disease subset, others develop potentially sight or life-threatening manifestations, due to ocular, neurological or major vascular involvement.3 The multiorgan involvement and the wide range of clinical spectrum often make the management of BS challenging; moreover, the relapsing course of the disease can determine exacerbations of symptoms over time4 and irreversible organ damage accrual.5 6 Various demographic factors, such as age at disease onset, duration of disease and gender, are considered predictive of poor outcomes in the short and long term. In fact, younger male patients have a more severe disease, leading to increased morbidity and mortality.7 Quality of life (QoL) is highly impaired in patients with BS, and this is related to different issues, including disease activity and severity.8–12 Although the frequency of ocular BS can vary according to the geographical area, the overall frequency of ocular involvement is around 50% of patients.3 Uveitis due to BS can be challenging to treat and even in those treated, a significant portion of patients experience loss of vision in the decade following the onset of the ocular symptoms.1 2 Neurological involvement, also known as neuro-BS, is characterised by parenchymal lesions or vascular events, with a prevalence that varies from 2% to 50%, thus representing an important cause of morbidity and mortality.2 Finally, vascular involvement, most frequently characterised by recurrent superficial and deep vein thrombosis, or arterial aneurysms, can occur in up to 50% of patients with BS, severely affecting the prognosis of BS.11
Besides the use of glucocorticoids (GC), other therapies are used to manage the systemic manifestations of BS, such as colchicine, azathioprine (AZA), cyclosporine A (CsA) and cyclophosphamide. Over the last two decades, the benefits of anti-tumour necrosis factor (TNF) alpha agents, either infliximab (IFX) and adalimumab (ADA) have been shown, not only for severe and refractory ocular disease, but also for treating almost all the systemic manifestations of BS12–23; accordingly, the European Alliance of Associations for Rheumatology (EULAR) recommendations for the management of BS,24 indicate that monoclonal anti-TNF antibodies should be considered in severe disease as first-line or in refractory patients. However, the lack of controlled evidence regarding anti-TNF use, still represents a main issue and these drugs are used as off-label therapy. This may also limit patients access to treatment in some countries.
The present trial aimed at evaluating the efficacy and safety of IFX or ADA in patients with BS with mucocutaneous manifestations, refractory to the standard of care (SoC) with AZA or CsA.
Methods
Study design and participants
This is a phase 3, multicentre, prospective, randomised, evaluator-blind, active-controlled, parallel-group study to evaluate the efficacy and safety of either IFX or ADA in patients with BS after induction therapy with systemic GC.
Patients were recruited from four tertiary referral centres for BS in Italy (Pisa, Florence, Siena and Cagliari). The trial was conducted in accordance with the Declaration of Helsinki (General Assembly of the World Medical Association 2014) and was approved by ethics committees at each participating centre. Written informed consent was obtained from all enrolled patients. Data were collected in accordance with Good Clinical Practice guidelines. Data of the study have been reported according to Consolidated Standards of Reporting Trials reporting guidelines.25
To be eligible, participants were required to be between 18 and 65 years old, diagnosed with BS according to the 1990 International Study Group criteria, presenting active mucocutaneous manifestations (presence of recurrent oral ulcers and/or genital ulcers and/or cutaneous manifestations), that have been occurred while on treatment with either AZA or CsA for at least 3 months prior to the study entry.
Key exclusion criteria were ‘end-stage’ BS, with severe retinal damage or central nervous system irreversible damage, visual acuity <1/10 in both eyes or bilateral permanent blindness; other severe BS manifestations, that is, arterial aneurysm, thrombosis of the caval, hepatic veins or cerebral sinuses, infection at screening or frequent acute or chronic infections within 3 months prior to the study entry, congestive heart failure, multiple sclerosis or any other central demyelinating disorder, history of malignancy within previous 5 years (except curatively excised skin cancer), organ transplantation (except cornea), substance abuse within 3 years, evidence of active or latent tuberculosis (TB), enrolment in other investigative clinical trial, prior history of anti-TNF alpha agents’ or other monoclonal antibody treatments, or known allergy to murine or chimeric proteins, hypersensitivity to the active substances or to any of the excipients, history of HIV, hepatitis C virus (HCV) or hepatitis B virus (HBV) infections, pregnancy or lactation were also considered exclusion criteria. Sex was self-reported as either male or female. Participants were approached by their usual care team and gave written informed consent before taking part in the study.
Randomisation and masking
Subjects recruited from the individual investigator’s clinical centre if fulfilling study entry criteria were randomised using a centralised computerised randomisation system. The investigator entered the subject number and key subject information into the background and demographic page and the system assigned the treatment for that subject according to a predefined computer-generated randomisation list which was blinded to each investigator. The computerised randomisation system considered block randomisation (1:1) within centres. Centralised blocked randomisation ensured that no selection bias was introduced when assigning the patients to study treatment. While patients and personnel who administered investigational medicinal product were open fashion, those investigators evaluating responses of ocular/neurological/mucocutaneous manifestations remained blinded with regard to the randomised treatment assignments.
Procedures
All subjects underwent screening evaluations to determine eligibility. The screening period had a duration of 90 days. All eligibility criteria must be met prior to week 0. The eligible subjects were randomised to receive either IFX (5 mg/kg intravenously administered at weeks 0, 2 and 6 and then every 6–8 weeks) or ADA (40 mg subcutaneously every 2 weeks) (ratio 1:1). In the event of relapse, patients were treated according to physicians’ local practice and the EULAR guidelines.24 All subjects were treated for 24 weeks and followed for 12 additional weeks after treatment was completed or discontinued.
Prior and concomitant therapies allowed were AZA (up to 2 mg/kg/day) or CsA (up to 5 mg/kg/day for the first month, to be tapered 0.5 mg/kg every 2 weeks until a maintenance dose of 3 mg/kg/day).
At the enrolment, high-medium doses of GC was permitted when required for the active disease and following this schedule: 6-methylprednisolone (0.5 g for body weight <50 kg or 1 g for body weight >50 kg intravenously for 3 days; then to taper to 40 mg/day intramuscularly for 3 days, then to reduce to 20 mg/day orally for 3 days, finally tapered of 4 mg/weekly until suspension). Traditional disease-modifying antirheumatic drugs and other biological drugs (except IFX and ADA) were not permitted. During the treatment period, the patients regularly underwent physician’s global assessment of disease activity (including Behçet’s Disease Current Activity Form (BDCAF) evaluation), physical examinations and measurement. During each visit, the investigators evaluated eligibility assessment, safety, efficacy, adherence to treatment and drug supplies. In the case of concomitant ocular involvement patients underwent at baseline visual acuity assessment, fundus oculi and tonometry monthly and fluorangiography and coherent radiation optical tomography at baseline and at the end of the treatment period. Similarly, in the case of neurological involvement, an MRI of the brain plus neurological examination was performed at study entry and at the end of the study period.
The study duration planned was 36 months, however due to the COVID-19 pandemic was extended for one further year to complete the analysis of the data.
Outcomes
The primary study outcome was the time to response of mucocutaneous manifestations over 6 months in anti-TNF alpha agents’ treatment, also evaluating the time to response of any concomitant vital organ involvement (mainly focusing on ocular and neurological involvement).
Patients were considered as ‘responders’ if obtaining the remission of mucocutaneous involvement (ie, the improvement of active manifestations and absence of new episodes of oro-genital ulcers and/or erythema nodosum) during a single-blind, complete treatment period with either IFX or ADA. Moreover, when concomitant ocular or neurological involvement were associated with the mucocutaneous manifestations, patients were considered responders in case of improvement of active manifestations and/or absence of new ocular attacks involving the posterior eye segment and in case of improvement of active manifestations and/or of the absence of new ischaemic pons-mesencephalon lesions and/or absence of meningoencephalitis with brainstem involvement, respectively.
Secondary outcomes were represented by the evaluation during all the treatment and follow-up period of: the proportion of BS subjects who had a relapse of mucocutaneous manifestations while on anti-TNF alpha agents (including also organ vital manifestations); time to relapse; disease activity measured by BDCAF; adherence to anti-TNF therapy defined as the administration/intake >70% of anti-TNF alpha agents in the considered period; the retention on anti-TNF alpha agents and reasons of withdrawal; effects on QoL evaluated by standardised questionnaire (ie, SF-36).
The safety and tolerability profiles of anti-TNF alpha agents were evaluated as the frequency of adverse events (AEs) and serious adverse events (SAEs) while on study. Patients were monitored for treatment-emergent AEs and SAEs every 2 weeks. Physical examinations and clinical laboratory tests were conducted at screening and every 4 and 8 weeks, respectively.
Statistical analysis
Sample size calculation for the study was based on the assumption that the safety population would be used for statistical analyses, therefore considering as population for analysis of all subjects who received at least one dose of study treatment. Moreover, response to anti-TNF alpha agents, that is, remission of mucocutaneous manifestations (and of any possible concomitant organ vital involvement) was evaluated at month 6. Due to the binomial nature of the outcome variable (0=non responder, 1=responder), we hypothesised that the use of the two anti-TNF-alpha agents could have made a difference of about 30% from baseline on the achievement of remission. With alpha error=0.05 and a power of 0.80, 23 patients per group were needed. The enrolled patients were followed for 18 months for clinical assessments and treatments. The efficacy variables were analysed in the population, that is, all subjects randomised to either IFX or ADA and exposed to at least one dose of the anti-TNF alpha agent as per random with at least one efficacy assessment since the treatment started. Maintenance on anti-TNF alpha agents was evaluated considering the number of days from treatment start to the discontinuation of treatment, which could lead to the switch to another therapy or the add-on of an immunosuppressant (IS) SoC drugs or to study withdrawal. Comparisons of qualitative data between the two groups were made by the χ2 test or the Fisher’s Exact test. Quantitative data were compared by means of the Student’s t-test for paired data or by the Wilcoxon’s test in case of non-normally distributed data. Time to response and time to relapse were evaluated as the number of days from treatment start to the event occurrence, and different groups treated with either IFX or ADA were compared by means of the log-rank test. A Cox model was defined to evaluate risk also adjusting for potential confounders. All analyses were performed using Stata V.16 and a p value<0.05 was considered for statistical significance.
Role of funding source
The funder of the study had no role in study design, data collection, data analysis, data interpretation or writing of the report.
Results
Between January 2018 and January 2020, a total of 42 patients with BS underwent a screening visit. Among them, 41 were considered eligible and included in the study: 19 were randomised to the ADA group and 22 to the IFX group. One patient discontinued the study after the randomisation because of being unavailable to continue the study (figure 1).

Consolidated Standards of Reporting Trials diagram. ADA, azathioprine; IFX, infliximab.
The baseline characteristics of the participants included in the study are shown in table 1.
Baseline characteristics of the population included in the study.
All but two patients (one in the IFX and one in the ADA group) completed the study as per-protocol. In detail, one patient in the IFX had hypersensitivity reaction type 1 and then discontinued, while one patient in the ADA group discontinued having received a diagnosis of myopathy at day 112. Accordingly, retention on treatment was 94% and 95% in ADA and in the IFX group, respectively.
As shown in table 2, we registered two AEs in the ADA group; only one of them may be considered as serious (myocardial infarction), although it was reported immediately after the last evaluation and in a subject with independent cardiovascular risk factors. In the IFX group we registered three AEs, all not serious.
AE and SAE in ADA e IFX groups
With respect to the overall evaluation of the efficacy (ie, the percentage of patients with mucocutaneous manifestations, even when these were associated with concomitant sight-threatening uveitis or neurological involvement) over 6-month anti-TNF alpha agents’ treatment, the primary outcome was met by 17 (94%) patients in the ADA group and 14 (64%) in the IFX group (p value=0.023) (figure 2). In detail, in the ADA group, one patient had relapse of mucocutaneous manifestations; as for the IFX group, one patient experienced anterior uveitis (P0015), P0022 had paresthesias and buccal rima deviation, P0035, P037 and P0020 had cutaneous manifestations. Moreover, as detailed above one additional patient exited the study. As far as mucocutaneous involvement is concerned, we did not find any difference according to isolated or associated mucocutaneous involvement to other kind of involvement.

Frequency distribution (%) of treatment responders and non-responders in the ADA and IFX group. ADA, adalimumab; IFX, infliximab.
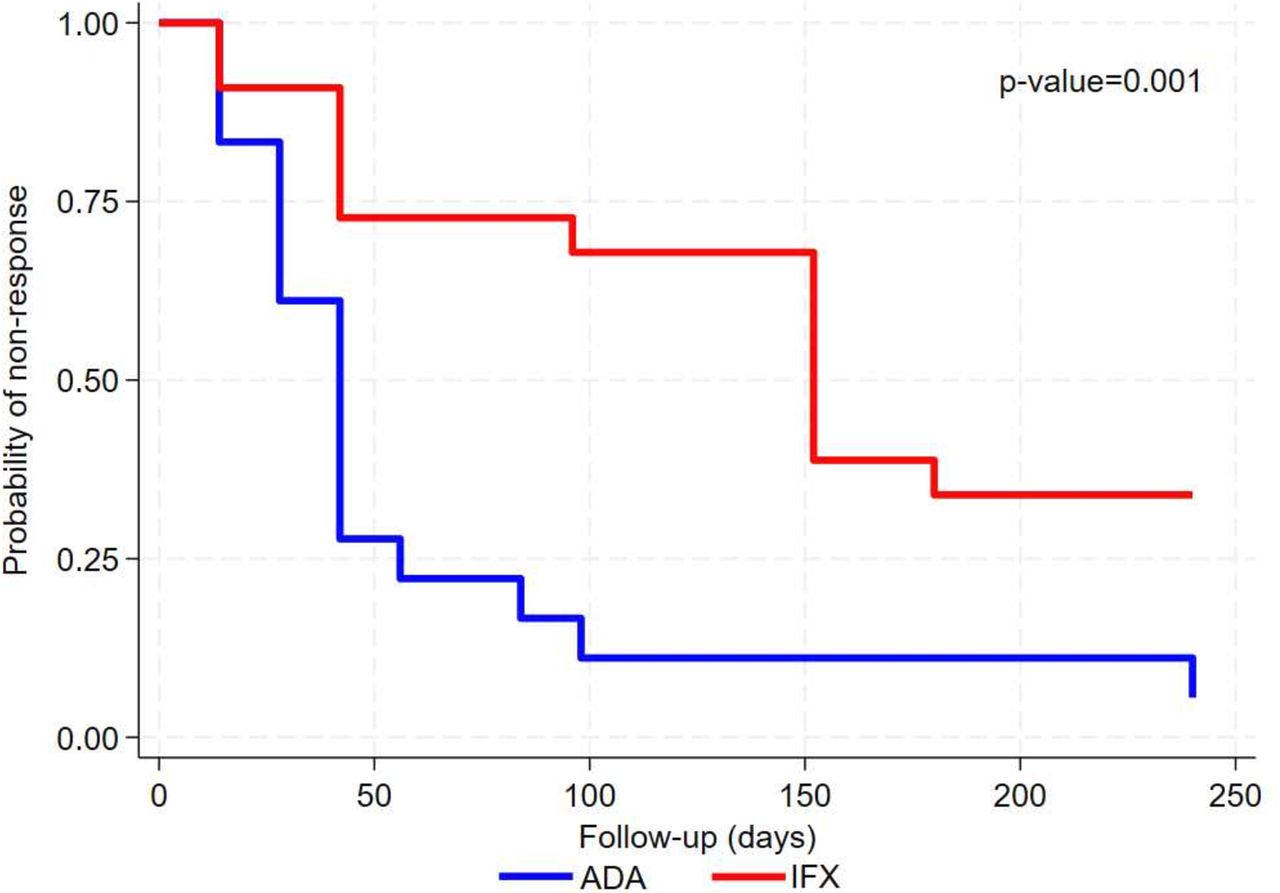
To what concern the primary outcome, time-to-response, resolution of sight-threatening uveitis and/or neurological and/or mucocutaneous manifestations was achieved significantly quicker in the ADA group as compared with the IFX group with a median time to response to the treatment being equal to 42 days and 152 days, respectively, p value=0.001 for log-rank test. Figure 3 shows the Kaplan-Meier curves for the risk of non-response in the two treatment groups.

Kaplan-Meier curves for the ADA and IFX group in terms of non-response to treatment (primary outcome). ADA, adalimumab; IFX, infliximab.
When analysing time to response considering a Cox model, significant differences were observed between the two groups with the risk of non-response being higher for the IFX group as compared with ADA, both at unadjusted, HR 2.56 (95% CI 1.22 to 5.26) p value=0.013, and adjusted analysis also controlling for variables resulted to be significantly different between groups (ie, smoking habits and GC use), HR 3.33 (95% CI 1.30 to 8.33), p value=0.012 for IFX versus ADA.
Considering both the randomisation visit and subsequent follow-up evaluation a total 5 (27.8%) patients in the ADA group and 9 (40.9%) patients in the IFX group experienced ocular, mucocutaneous or neurological manifestations (p value=0.510). In detail, 4 (22.2%) patients in the ADA and 5 (22.7%) patients in the IFX group have active manifestations (one or more) at day 0 (randomisation visit), p value=1.000.
A disease flare was observed in a minority of patients in the ADA (2/18, 11.1%) and in the IFX group (7/22, 31.8%) (p value=0.149). Tables 3 and 4 show the number of relapses per visit for each of the two drugs.
Number and rate of relapses per visit in the adalimumab group
Number and rate of relapses per visit in the infliximab group
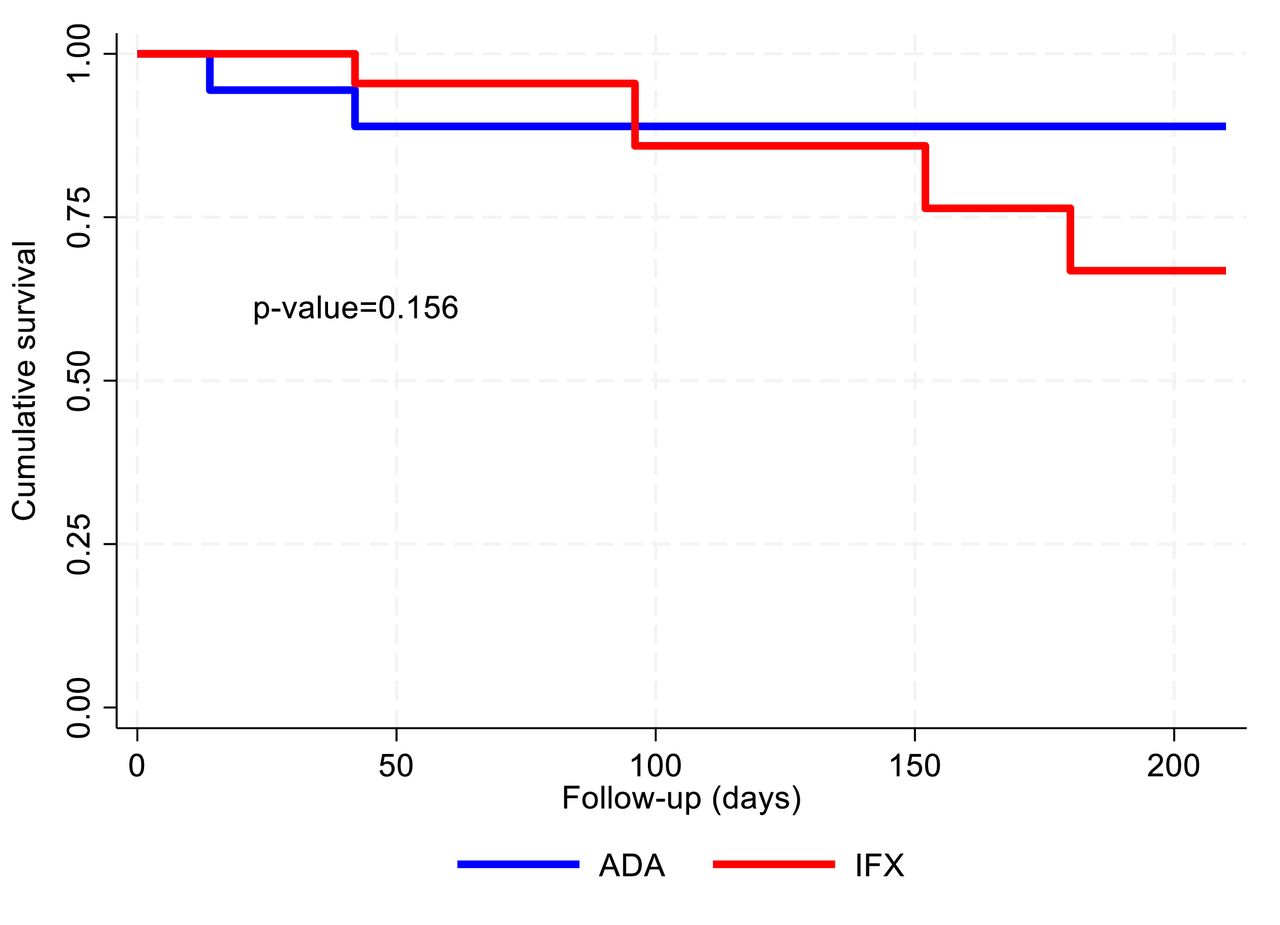
A number of relapses shown in the table exceeds the number of patients having manifestations reported above as some patients experienced multiple manifestations. In detail, in the ADA group, one patient experienced a manifestation on day 42 and another patient experienced a manifestation on days 14 and 42, the same manifestation was present also on day 0 and resolved after day 42. In the IFX group. five patients experienced single manifestation during the study follow-up while the others showed multiple manifestations (P003: days 96 and 152, P0035: days 152 and 206). Moreover, considering the time to first relapse, figure 4 shows the Kaplan-Meier curves for the ADA and IFX groups, no differences were observed between the two groups (p value=0.156 for log-rank test).

Kaplan-Meier curves for the ADA and IFX group in terms of disease relapses. ADA, adalimumab; IFX, infliximab.
The simple Cox model suggested no significant differences in the risk of relapse between the two groups, HR 2.92 (95% CI 0.61 to 14.04), p value=0.182 for IFX versus ADA, when adjusting for variables resulted to be significantly different between groups (ie, smoking habits and GC use) a significantly higher risk of relapse was estimated in the IFX group as compared with ADA, HR 7.57 (95% CI 1.14 to 50.19), p value=0.036.
Adding more details, five patients experienced ocular manifestations in the IFX group and had no active sign at baseline, none of the patients in the ADA group that had no active ocular manifestations at baseline experienced them during follow-up. One patient in the ADA and another one in the IFX group experienced new neurological manifestations during follow-up. Incidence of new mucocutaneous manifestations were observed just for one patient in the IFX group.
All the dimensions of QoL as assessed by SF-36 resulted to be significantly improved (from baseline) at the last visit, physical function (p value=0.005 and 0.001 in the ADA and IFX group, respectively), role limitations due to physical health (p value=0.004 and 0.001 in the ADA and IFX group, respectively), role limitations due to emotional problems (p value=0.004 and 0.001 in the ADA and IFX group, respectively), energy/fatigue (p value=0.007 and 0.001 in the ADA and IFX group, respectively), emotional well-being (p value=0.007 and 0.001 in the ADA and IFX group, respectively), social functioning (p value=0.001 both in the ADA and IFX group, respectively), pain (p value=0.001 both in the ADA and IFX group, respectively), general health (p value=0.002 and 0.003 in the ADA and IFX group, respectively), health change (p value=0.001 both in the ADA and IFX group, respectively). QoL at the end of the study remained similar between the two groups (figure 5, tables 5 and 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Radar plot for QoL (SF-36) scores at baseline and follow-up in the ADA and IFX group. Dashed lines refer to baseline values while dotted lines represent follow-up values, in both groups QoL significantly increased for all the dimensions considered. ADA, adalimumab; EF, energy/fatigue; EW, emotional well-being; GH, general health; HC, health change; IFX, infliximab; PA, pain; PF, physical functioning; QoL, quality of life; RE, role limitations due to emotional problems; RP, role limitations due to physical health; SF, social functioning; SF-36, Short Form Health Survey 36.
Quality of life (SF-36) among the patients included at baseline
Quality of life (SF-36) in the two arms after the study period
Also, disease activity as evaluated by the BDCAF significantly improved (from baseline) at the end of the study period both in the ADA and in the IFX group (p value<0.001 for both) without significant differences between group both at baseline and at the end of the study (table 7). Moreover, we did not observe differences in terms of other concomitant therapies, GC tapering in those cases characterised by major organ involvement. Table 8 summarises the medium dose of GC, the medium number of oral ulcers, the Physician Global Assessment and the Patient Global Assessment at baseline and at the end of the follow-up period.
Behçet’s Disease Current Activity Form in the two arms at baseline and at the end of follow-up
Medium dose of GC, the medium number of oral ulcers (OU), the Physician Global Assessment and the Patient Global Assessment at baseline and at the end of the follow-up period
When considering exclusively mucocutaneous involvement, the percentage of patient responders over 6-month anti-TNF alpha agents’ treatment was similar between groups being 100% in the ADA group and 86.4% in the IFX group (p value=0.238) (online supplemental figure 1). On the other hand, to what concern the primary outcome, time-to-response, resolution of mucocutaneous manifestations was achieved significantly quicker in the ADA group as compared with the IFX group, p value=0.002 for log-rank test. (Online supplemental figure 2) shows the Kaplan-Meier curves for the risk of non-response in the two treatment groups.
Supplemental material
Supplemental material
Globally, the treatment with both IFX and ADA were well tolerated by the patients. In terms of self-reported adherence to treatment, 100% of IFX patients were completely adherent to the therapy during the study period. In the group of ADA, only one patient reported to skip the therapy (one time over the study period).
Discussion
The overall results of this study report on the efficacy and safety of both ADA and IFX in achieving remission in patients affected by mucocutaneous involvement due to BS refractory to traditional immunosuppressive therapy. These findings agree with data already published14–24 26 and also with the common real-life experience on the use of anti-TNF alpha drugs, in managing several types of organ involvement in BS.
To our knowledge, this is the first controlled study assessing in the same study the efficacy and safety of both ADA and IFX. Indeed, collecting further evidence on the effectiveness of both drugs in BS can be considered an important milestone also for encouraging the health authorities to speed-up the procedures for approving their on-label use. In our study, no significant differences were found with respect to the efficacy of anti-TNF used as monotherapy or in association with an immunosuppressive agent. This is in line with data coming from a previous multicentre study from the French Behçet Network, which analysed a large series of patients with BD with ocular and extraocular manifestations of BD treated with anti-TNF agents and showing that IFX and ADA seem to have similar efficacy and safety profile, without significant differences respect to the efficacy of anti-TNF therapy used as monotherapy or in association with an immunosuppressive agent.27 Similarly, both IFX and ADA were reported as efficacious in improving uveitis macular oedema in patients with BD in a Japanese retrospective study,28 while other literature data have reached similar conclusions while showing a slight difference between IFX and ADA, being ADA to be associated with better outcomes than IFX after 1 year of follow-up.29
In our study, both ADA and IFX showed excellent retention on treatment, and safety profile. In terms of efficacy, the occurrence of mucocutaneous manifestations during follow-up, that were defined as relapses, were seen in a minority of patients, both in the ADA and in the IFX group, as well as ocular or neurological involvement. However, on the basis of both time-to-response and the percentage of responders in the two groups, the data seem apparently to indicate a slightly higher efficacy profile of ADA compared with IFX. Nevertheless, the half-life of the molecules is different, and this might have affected the evaluation of time to response in the IFX group and can probably explain an earlier response to ADA treatment rather than a higher efficacy, also due to the study period of observations of 6 months.
Also, all the dimensions of QoL as assessed by SF-36 resulted to be significantly improved in both groups from baseline, as well as the assessment of disease performed through the BDCAF. One of the main characteristics of BS is to have a variable course with relapsing and remitting phases, as well as higher disease activity, especially in the first years after disease onset.26 30–33 The complexity of BS phenotype variability requires a multidisciplinary view and a customisation of the therapeutic approach to the individual patient is crucial in order to improve the overall disease prognosis and the QoL of both the patient and the caregivers/family members.33 34 Different factors can have a worse impact on the prognosis, including the type of organ involvement, early disease onset and male sex; however, also manifestations of benign course, such as mucocutaneous signs and symptoms, can definitely affect QoL of patients with BS. All these considerations highlight how important is to start the most appropriate treatment early in the disease.30–35
The main limitations of this study include the small sample size and the peculiarity of the study design (ie, AZA and cyclosporin A (Cy A)) CyA failure vs use of IFX or ADA), which at the same time represents an aspect that traces pragmatically what happens in clinical practice. However, considering the rare prevalence of BS, conducting a trial of a large-scale is not always feasible and in light of the inherent characteristics of small populations, we can accept even low numbers of case studies. In addition, the study design, although peculiar, may reflect frequent treatment scenarios in real life, that is, the choice to use anti-TNF alpha agents in case of failure or partial response to traditional immunosuppressive treatments and this represents a main point of strength of the study. Another relevant aspect is related to the multicentre nature of the population enrolled, that is, supposed to be truly representative of the disease patterns. Moreover, we should take into account that patients with BS enrolled in this study were by definition refractory to the SoC therapy and this scenario is quite similar to what happens when managing patients with BS. The importance of this study can therefore also be attributed to the fact that a critical issue often experienced in real life has been answered by means of an randomised controlled trial (RCT) in a small population. Besides these considerations, an additional added value of the study is that it assessed adherence to therapy, which was excellent, being about 100%.
In summary, ADA and IFX were generally well-tolerated and efficacious in patients with BS who showed an inadequate response to prior treatments with at least AZA or CyA; the efficacy was particularly prominent in the subanalysis of only mucocutaneous patients, similarly to previous data observed in a controlled study on etanercept.36 In 2016, ADA received European Medicine Agency approval for the treatment of non-infectious uveitis and panuveitis; besides the recommendation of considering ADA in these cases, the data from our study highlight that both drugs demonstrated robust improvement in clinical response, QoL, besides a high level of adherence to treatment and tolerability in different kinds of organ involvement. Although a more detailed treat-to-target profile is yet to be better defined,37 these results are also crucial in terms of prescriptiveness (currently off-label), not only in Italy but also beyond national borders, as the evidence coming from real life still needs to be confirmed by growing data from clinical trials.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Northwest Vast Area Ethics Committee (CEAVNO) for Clinical Trials Protocol Number: FARM 12LTAT, EudraCT Code 2017-000845-39. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors wish to thank the Italian Patients’ association for Behçet’s disease (SIMBA OdV) that has encouraged the creation of this study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Josef S Smolen
X @SaraTalarico2
Deceased 2015
Contributors RT conceived the paper; RT and ODCA were involved in the study design. VL and AG performed statistical analyses of the data. RT and NI wrote the draft of the manuscript. All authors were involved in the discussion and interpretation of the results. All authors repeatedly edited the manuscript and approved the final version. RT and MM are the guarantors.
Funding This study was financed through a grant from the Italian Medicines Agency (AIFA) Protocol Number: FARM12LTAT.
Competing interests None declared.
Patient and public involvement Patient and public were not directly involved in any stage of the study; however, the Italian Patients’ association for Behçet’s disease (SIMBA OdV) has indirectly encouraged the creation of this study by highlighting, in several patients’ conferences, the need of having more evidence from clinical trials regarding the use of the two drugs studied and the need to approve a potential on-label use of them.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.