Article Text

Abstract
Objectives This work aimed to resolve the conflicting reports on Prevotellaceae abundance in the development of rheumatoid arthritis (RA) and to observe structural, functional and temporal changes in the gut microbiome in RA progressors versus non-progressors.
Methods Individuals at risk of RA were defined by the presence of anticyclic citrullinated protein (anti-CCP) antibodies and new musculoskeletal symptoms without clinical synovitis. Baseline sampling included 124 participants (30 progressed to RA), with longitudinal sampling of 19 participants (5 progressed to RA) over 15 months at five timepoints. Gut microbiome taxonomic alterations were investigated using 16S rRNA amplicon sequencing and confirmed with shotgun metagenomic DNA sequencing on 49 samples.
Results At baseline, CCP+ at risk progressors showed significant differences in Prevotellaceae abundance compared with non-progressors, contingent on intrinsic RA risk factors and time to progression. Longitudinal sampling revealed gut microbiome instability in progressors 10 months before RA onset, a phenomenon absent in non-progressors. This may indicate a late microbial shift before RA onset, with Prevotellaceae contributing but not dominating these changes. Structural changes in the gut microbiome during arthritis development were associated with increased amino acid metabolism.
Conclusion These data suggest conflicting reports on Prevotellaceae overabundance are likely due to sampling within a heterogeneous population along a dynamic disease spectrum, with certain Prevotellaceae strains/clades possibly contributing to the establishment and/or progression of RA. Gut microbiome changes in RA may appear at the transition to clinical arthritis as a late manifestation, and it remains unclear whether they represent a primary or secondary phenomenon.
- Rheumatoid Arthritis
- Anti-Citrullinated Protein Antibodies
- Risk Factors
Data availability statement
Raw sequencing data available upon reasonable request from the corresponding author.
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Individuals with rheumatoid arthritis (RA) and those at risk possess distinct gut microbiomes when compared with healthy controls; however, detailed insights into the temporal and compositional changes in the gut microbiome as individuals progress to RA are lacking.
Prevotellaceae, and particularly Prevotella copri, have been inconsistently associated with RA development, and while early studies showed an overabundance more recent cohorts that question these observations have emerged.
WHAT THIS STUDY ADDS
We identified a phenomenon where specific strains/clades of Prevotellaceae are enriched, while others are depleted, depending on an individual’s risk profile and time to arthritis progression.
Anticyclic citrullinated protein antibody-positive individuals at risk of RA are characterised by a low-diversity gut microbiome that may become more unstable in the 10 months preceding the transition from preclinical RA to established disease (ie, the development of clinical arthritis).
This instability is marked by accumulation of multiple bacteria, including but not limited to Prevotellaceae.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
These findings illustrate the dynamic nature of the gut microbiome, reflecting an individual’s underlying risk profile and transition from being at risk to developing clinical arthritis.
This evolving understanding could significantly impact approaches to RA prediction, prevention and personalised treatment strategies, emphasising the need for further research into microbiome-based diagnostics and therapeutics.
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disorder affecting over half a million individuals in the UK. The hallmark of RA is progressive joint disease, with potential for systemic involvement. Understanding the RA disease spectrum with recognition of at risk individuals has propelled RA research into prevention strategies. The generation of IgA class anticitrullinated protein antibodies (ACPAs) in individuals at risk of RA,1 combined with epidemiological links with smoking2 and periodontal disease,3 points to a mucosal origin of inflammation. The mucosal origin hypothesis proposes that localised inflammation at mucosal sites can initiate a broader immune response,1 via T cell activation and a subsequent inflammatory cytokine cascade, leading to B cell antibody production. Supporting this, an immunoglobulin class switch from IgA ACPAs to IgG ACPAs4 5 indicates potential triggering of systemic autoimmunity by diverse antigenic stimuli at mucosal sites. This shift, accompanied with broadening of antibody targets,6 7 suggests that mucosal barrier deterioration and the ensuing spread of an IgG ACPA response might be more significant in the initial stages of RA than the loss of tolerance to self-antigens.
Profiling of the gut microbiome in individuals at risk of RA and people diagnosed with RA consistently demonstrates a dysbiotic microbiome when compared with healthy controls.8–21 However, there remains little consensus on the bacterial constituent members of an RA-related dysbiosis. Subsequently, a variety of gut bacteria have been implicated as a potential impetus in the development of RA, none more so than Prevotella copri. Prevotella species have been demonstrated to be overabundant in new-onset rheumatoid arthritis (NORA),9 11 22 in at risk individuals8 and especially those with genetic risk.10 Their abundance decreases after disease-modifying antirheumatic drug (DMARD) therapy, with reversion to a eubiotic state on treatment.22 Furthermore, mouse models support a role for Prevotellaceae strains derived from patients with RA in RA development.23 However, Prevotellaceae overabundance does not appear to be an ubiquitous finding across all RA gut microbiome studies,16–21 including our own previous work.24 Additionally, researchers have failed to replicate their findings of Prevotellaceae overabundance in larger cohorts.25 Increasingly, numerous other bacteria have also been associated with RA development, including Subdoligranulum,26 Lactobacillus,11 20 Bacteroides species14 17 and Streptococcaceae,20 21 among others.
EULAR, the European Alliance of Associations for Rheumatology, defines at risk populations for RA based on a combination of genetic, environmental and immunological factors that predispose individuals to RA.27 Therefore, heterogeneous RA at risk populations and confounding within microbiome studies further complicate the RA gut microbiome debate. This work examines a comprehensively phenotyped cohort of anticyclic citrullinated protein-positive (anti-CCP+) at risk individuals without clinical arthritis. We combined cross-sectional and longitudinal investigations of the gut microbiome with extensive clinical data, adjusting for potential confounding, to uncover microbial associations in the early RA continuum, with particular attention to Prevotellaceae strains.
Methods
Patient and public involvement statement
Before starting this research, patients contributed to the study’s design and feasibility, influencing recruitment, sample return and dissemination of outputs through a patient-focused discussion group at Chapel Allerton Hospital (CAH), Leeds. During the study, participant feedback on the stool collection kit was gathered and led to its redesign.
Study design and participants
This study used both cross-sectional and longitudinal approaches, involving participants from three groups: CCP+ at risk (n=124), newly diagnosed RA (NORA, n=7) and healthy controls (n=22). The study aimed to differentiate gut microbiome bacterial shifts due to confounding factors from those associated with RA progression, using comprehensive phenotypic data and serial stool samples as CCP+ at risk individuals progressed towards arthritis development.
The research was conducted at CAH, Leeds, UK. Individuals at risk of RA were selected from the Leeds CCP+ at risk cohort, as previously outlined.28 These participants were recruited nationally from both primary and secondary care settings, presenting with recent musculoskeletal pain (onset <3 months) and positive for anti-CCP antibodies (CCP2 BioPlex 2200 Kit >2.99 IU/mL), but with no evidence of clinical synovitis/arthritis.
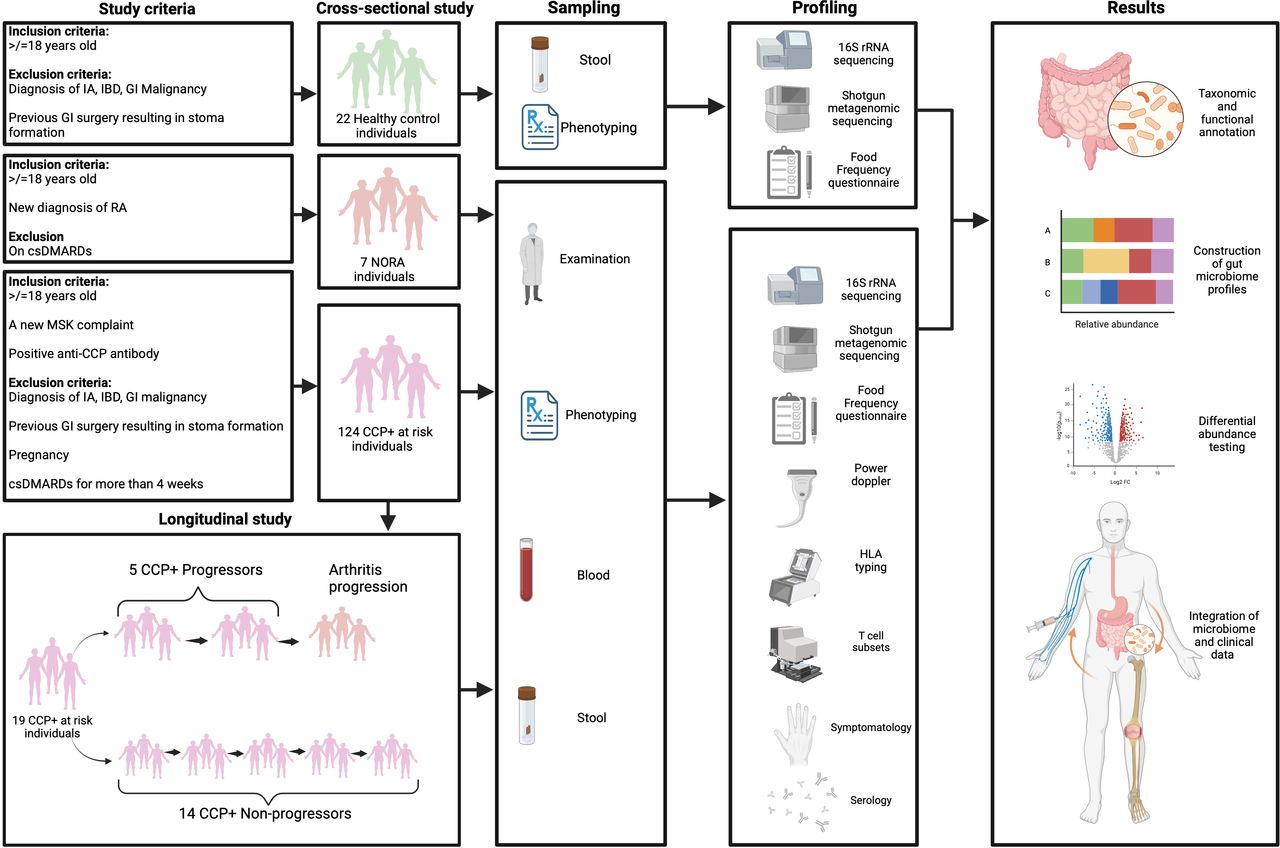
Within the CCP research clinic at CAH, individuals undergo longitudinal assessment for RA progression. An individual’s risk of arthritis development was calculated using a validated RA risk severity score, based on symptoms, joint ultrasound, human leucocyte antigen (HLA) status and antibody titre,28 enabling comparison of gut microbiomes across varying RA risk. Comprehensive data on demographics, medical history, medications, diet, bowel habit and lifestyle were also collected (figure 1).

Study design and methodological flow chart. The figure depicts the recruitment of healthy controls, CCP+ at risk individuals and those with NORA, with corresponding criteria and subsequent sequential steps from sampling through molecular profiling and data analysis. Incorporates stool and blood analyses, sequencing methods and clinical assessments, culminating in the evaluation of microbial composition and its correlation with RA progression. CCP, cyclic citrullinated protein; csDMARDs, conventional synthetic disease-modifying antirheumatic drugs; GI, gastrointestinal; HLA, human leucocyte antigen; IA, inflammatory arthritis; IBD, inflammatory bowel disease; Log2 FC; Log2 fold change; MSK, musculoskeletal; NORA, new-onset rheumatoid arthritis; RA, rheumatoid arthritis.
As a comparison, individuals with NORA were recruited from general rheumatology clinics at CAH. Those diagnosed with RA and meeting the EULAR criteria, irrespective of CCP antibody status, were invited to participate in the cross-sectional study as participants with NORA. These individuals provided a single stool sample within 48 hours of their RA diagnosis, prior to starting DMARD therapy. Local healthy controls, free of musculoskeletal symptoms and representing diverse socioeconomic backgrounds, were also included, providing a single stool sample for the cross-sectional study. These controls were approximately matched with the CCP+ at risk cohort in terms of age, gender and smoking status.
Assessments
CCP+ at risk individuals underwent a baseline assessment, followed by quarterly follow-up appointments for the first year, with annual visits thereafter. The study endpoint was the development of clinical arthritis. A research helpline enabled prompt reporting of symptom changes for timely clinical evaluation. At baseline, demographic, medical, drug, social, stool and dietary histories29 were collected. Clinical assessments at each visit included symptom review and joint assessment (78-joint count for tenderness and a 44-joint count for swelling).
Blood samples were collected at each visit for antibody testing, with the BioPlex 2200 machine used for anti-CCP2 testing and nephelometry for rheumatoid factor (RF). During the course of the study, a small number (n=11) of low titre CCP+ individuals transitioned to a negative CCP+ titre at the time of gut microbiome sampling. High-resolution joint ultrasonography using power Doppler (PD) was performed at baseline, 6 monthly for the first year, then annually thereafter and intermittently on symptom exacerbation. PD assessment included scanning of 38 joints bilaterally (metacarpophalangeal, proximal interphalangeal and metatarsophalangeal joints). HLA-DR (Human Leukocyte Antigen-DR isotype) typing occurred during the baseline visit. T cell subpopulations were quantified using flow cytometry, adjusting counts for age. T cell counts were taken at baseline, 6 months and then annually.
Participants were given in-house stool collection kits for home sample collection, returning the kit at their subsequent clinic visit. Samples were kept at room temperature at home before being frozen at −80°C for long-term storage.
Molecular and bioinformatic methods
DNA was extracted from stool samples using the QIAamp DNA Stool Mini Kit (Qiagen, Germany). The University of Leeds’ Next Generation Sequencing Facility at St James’s University Hospital campus conducted sequencing for both 16S rRNA amplicon and shotgun metagenomic analysis. For 16S rRNA sequencing, the V4 region was amplified with specific primers and sequenced using Illumina MiSeq 2500, generating 2×250 base pair (bp) paired-end reads. Data were analysed using the DADA2 method30 and SILVA reference database31 within the QIIME232 framework.
Shotgun metagenomic libraries were prepared with the Nextera XT DNA Library Prep Kit and sequenced on Illumina NextSeq 2000 using a 2×150 bp paired-end approach. The bioBakery workflow33 was used for taxonomic and functional analysis of the shotgun sequencing data (see online supplemental materials, pages 1–3 for additional details).
Supplemental material
All samples underwent 16S rRNA amplicon sequencing. In addition, 49 baseline samples, including CCP+ at risk individuals (n=27), individuals with NORA (n=7) and healthy controls (n=15), also underwent shotgun sequencing. This dual approach was employed to validate the findings from the differential analysis of the 16S data, particularly to confirm the observed trends in Prevotella abundance and to explore any inferred functional differences arising from altered microbiome composition.
Analysis
Cross-sectional
In both sequencing methodologies, analyses were conducted in R (V.3.6.2).34 Diversity was calculated using phyloseq35 and vegan36 packages. For the 16S approach, a rooted phylogenetic tree was generated in QIIME2,32 with alpha and beta diversity metrics calculated and Benjamini-Hochberg (BH)-adjusted p values (<0.05) determined via pairwise Wilcoxon rank-sum tests.
Permutational multivariate analysis of variance (MANOVA) was performed to assess the impact of metadata variables on beta diversity using three ecological metrics: Bray-Curtis, logged Bray-Curtis (Bray-Curtis was applied to scalar normalised counts (SNC) and logged data via the formula log10(SNC+10−6)+6) and unweighted UniFrac. Logged Bray-Curtis was used as an attempt to curtail outlying samples which might artificially amplify a signal within the Bray-Curtis index. Significant variables (p<0.05) were ranked by effect size and incorporated into a multivariate model to calculate cumulative variance explained.
For the shotgun method, a phyloseq object35 was constructed using the enzyme commission number data set, and principal component analysis was performed to assess functional diversity of the microbiome. MaAsLin237 was used for functional pathway analysis on prenormalised MetaCyc38 annotated pathway abundance files, using an false discovery rate (FDR) of <0.05 as a significance threshold.
Differential bacterial abundance between clinically relevant groups was investigated across both sequencing methods using DESeq2,39 adjusted for age and sequencing run. BLAST analyses supported further taxonomic annotation of significant Prevotellaceae strains. Differential abundance testing was performed at the genus level to determine higher taxonomic changes comparable to previous studies and at the strain level data to infer potential strain/subspecies information. In both methodologies, ggplot240 facilitated data visualisation. Only taxa reaching a BH-adjusted p value of <0.05 were labelled. Prevotella strains of interest were identified based on Prevotella strains that reached a p value of <0.05 in the DESeq2 models from the cross-sectional study. The Prevotella strains of interest from the DESeq2 models and their associations were plotted using a chord plot.
Longitudinal
Longitudinal gut microbiome changes were analysed using 16S rRNA amplicon sequencing. Pairwise Bray-Curtis dissimilarities were constructed, and comparisons were limited to within-participant timepoints. These pairwise timepoints were categorised based on the time elapsed from RA diagnosis for progressors or from baseline for non-progressors. A logged Bray-Curtis distance matrix was used to construct a hierarchical tree using the ‘dendrogram’ and ‘hclust’ functions, applying the ‘ward.D2’ clustering algorithm. Mixed-effects linear models were created using the MaAsLin237 package. Participants were assigned as the random-effect source in all models. To explore bacterial taxa, a centred log-ratio transformed data set was used at both the genus and strain levels.
Results
The cross-sectional study encompassed 153 participants; their characteristics are detailed in table 1. In the RA at risk cohort, 70% were female, with a median anti-CCP antibody titre of 73 IU/mL. During the study period, 30 individuals progressed to RA, with a median interval of 9.6 months between stool collection and RA progression. The clinical attributes of progressors, non-progressors and NORA cohorts are outlined in table 2. Microbiome summary statistics are available in the online supplemental materials, page 4-5.
Characteristics of the cross-sectional study participants
Baseline clinical characteristics of the cross-sectional musculoskeletal cohorts
CCP+ at risk individuals are characterised by a low diversity state, which is associated with CCP antibody titre, RF positivity and HLA shared epitope status
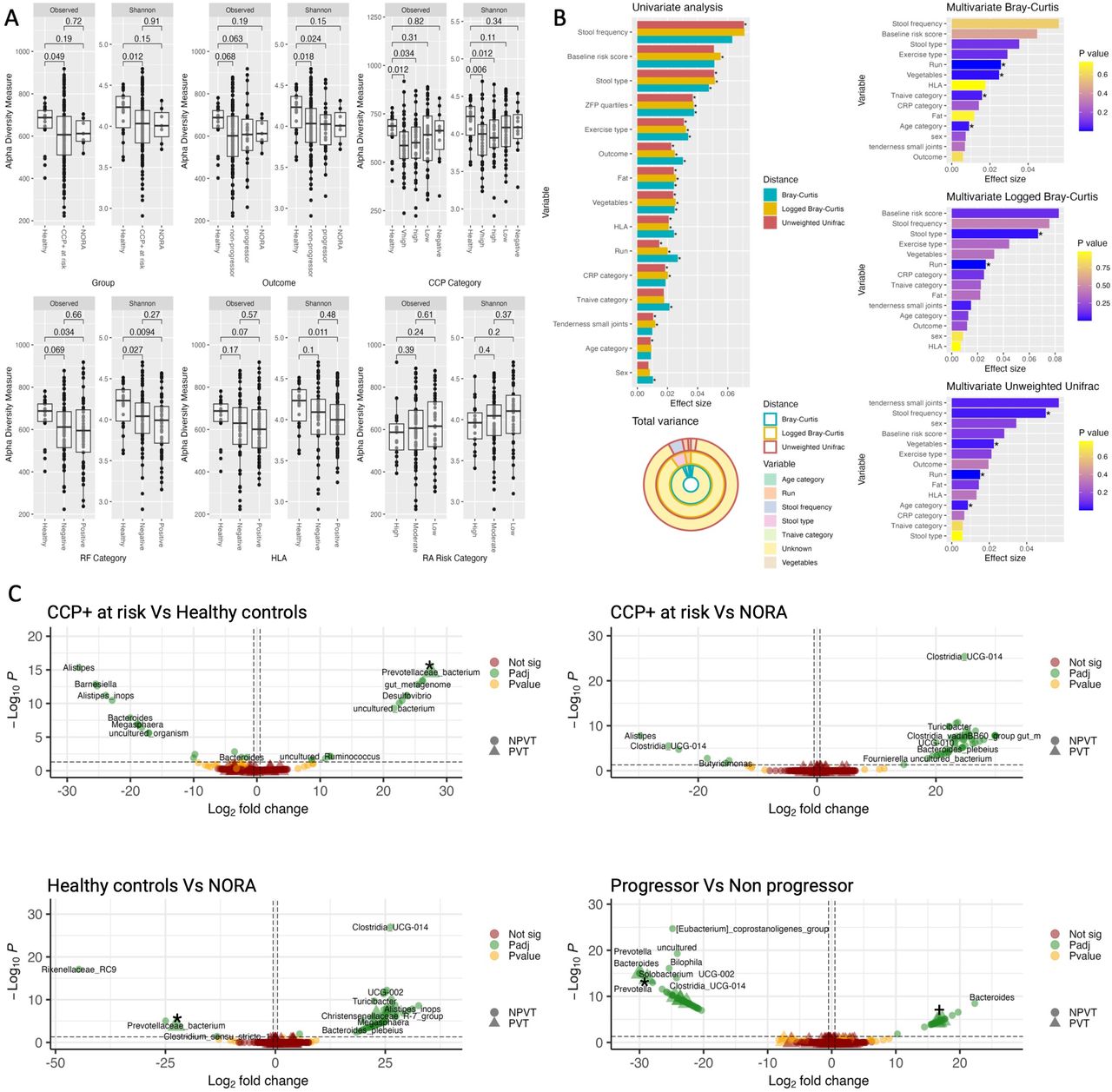
Shannon diversity was notably reduced among CCP+ at risk individuals compared with healthy controls (Wilcoxon, p=0.012; figure 2A). Diversity was reduced in both progressors and non-progressors, and appeared stratified by anti-CCP titre levels (figure 2A). Conversely, individuals with low or negative anti-CCP titres maintained diversity levels comparable to healthy controls. Both RF and HLA shared epitope (SE) positivity, known risk factors for arthritis development, were significantly linked to diminished diversity, as was steroid use (see online supplemental material, page 6). When examining the effects of these variables collectively in a multivariate model, none reached statistical significance (see online supplemental materials, page 6), suggesting that the individual contributions of these factors are less clear when considering the combined influence of all variables. Alpha diversity for measured lifestyle factors is presented in the online supplemental materials, page 6. Linear regression exploring variable effects on alpha diversity is presented in online supplemental materials, page 6-8.

Gut microbiome diversity, variance and differential abundance from 16S rRNA amplicon sequencing. (A) Cross-sectional alpha diversity plots. Boxplots of alpha diversity as measured using observed and Shannon diversity metrics. BH-adjusted p values generated by pairwise Wilcoxon rank-sum test. (B) Microbial variance plots. Univariate analysis of significant (p<0.05) metadata variables using permutational ANOVA on three measures of beta diversity: Bray-Curtis, logged Bray-Curtis and unweighted UniFrac. Variables ordered by decreasing effect size. Total variance ring plot demonstrating the total known and unknown variance present within the gut microbiome. Individual multivariate permutational ANOVA, adjusted for significant variables identified in the univariate models, with significance reached in at least two ecological distances. Variables ordered by effect size. (C) Differentially abundant taxa between CCP+ at risk, healthy controls and NORA and between progressors and non-progressors. Pairwise volcano plots of plotting log2 fold change against −log10 p value, coloured according to significance. Vertical line denotes adjusted p value threshold; horizontal line denotes 0.5 log2 fold change. The shape denotes PVT or NPVT taxa. Taxonomic labels were generated to give the lowest taxonomic rank available in SILVA. *Denotes ASV2058. +Denotes ASV1867. (B) Outcome refers to progressor, non-progressor, NORA and healthy individuals. Run refers to sequencing run. ANOVA, analysis of variance; BH, Benjamini-Hochberg; CCP, cyclic citrullinated protein; CRP, C reactive protein; HLA, human leucocyte antigen; NORA, new-onset rheumatoid arthritis; NPVT, not Prevotellaceae; PVT, Prevotellaceae; RA, rheumatoid arthritis; RF, rheumatoid factor; ZFP, zonulin family peptide.
Beta diversity analysis with respect to RA outcome status indicated no association between progression and enterotype (see online supplemental materials, page 9). Participants clustered according to dominant genera, primarily differentiating between Prevotella-dominated and Bacteroides-dominated microbiota. Progressors displayed both Prevotella and non-Prevotella predominant gut microbiome profiles. The analysis of 49 metadata variables using permutational analysis of variance (online supplemental materials, page 10-13) identified 15 variables with statistical significance (p<0.05) in at least one ecological metric (figure 2B), and subsequent multivariate models revealed sequencing run, age and vegetable intake as consistently significant factors across all three ecological distances, explaining microbial variance ranging from 8% to 10% (figure 2B). Alpha and beta diversity was calculated using 16S rRNA amplicon sequencing.
Progression from the at risk phase to arthritis development is associated with strain-specific changes in Prevotellaceae
A specific Prevotellaceae strain, ASV2058 (likely P. copri; BLAST analysis can be found in online supplemental materials, page 30-31), was enriched in the at risk population compared with healthy controls. This same strain (ASV2058) was also found to be overabundant in individuals with NORA compared with healthy controls, with no significant difference in this putative P. copri strain between at risk and NORA (16S rRNA amplicon sequencing; see figure 2C).
Models were constructed to investigate a baseline RA progression signature within the CCP+ at risk cohort by comparing progressors with non-progressors. Strain-level analysis demonstrates concordance with genus-level results (see online supplemental materials, page 14). Strain-specific phenomena are noted with both enrichment (three strains) and depletion (five strains) of Prevotellaceae-specific strains associated with progression (16S rRNA amplicon sequencing; see figure 2C). ASV2058 is included as one of those five depleted strains.
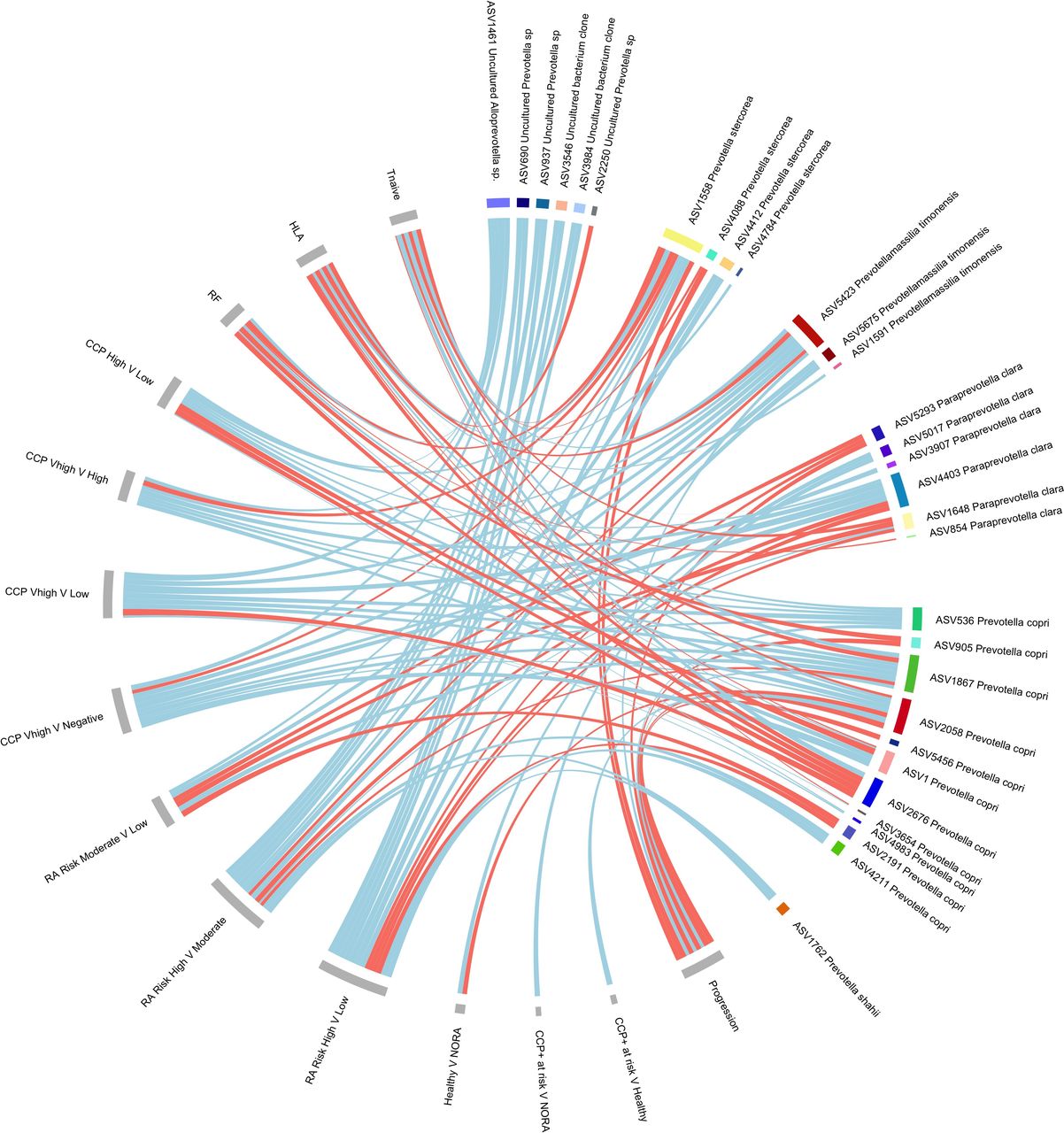
A phenomenon of strain-specific enrichment and concomitant depletion of Prevotellaceae strains was identified across multiple factors associated with higher risk of arthritis development, including anti-CCP antibody titre, RF positivity, HLA SE positivity, early immune dysregulation (T-naïve cells) and RA risk score (see online supplemental materials, page 15-19). All Prevotellaceae reaching nominal significance are detailed in online supplemental materials, page 20-29. Figure 3 provides a summary of Prevotellaceae strains found to be increased or decreased within the RA at risk population. Table 3 shows the proportion of each group containing Prevotellaceae and the proportion of Prevotellaceae strains of interest per group. From figure 3, Prevotellaceae strains ASV2058 and ASV1867 both had eight connections. RA risk category showed the highest number of associations with Prevotellaceae. Among all Prevotellaceae, putative P. copri strains had the most associations with RA clinical variables; however, other strains were also implicated, such as Alloprevotella, Paraprevotella clara, Prevotella stercorea, Prevotellamassilia timonensis and Prevotella shahii.

Prevotellaceae chord plot. Associations of Prevotellaceae strains reaching nominal significance from adjusted DESeq2 models from 16S rRNA amplicon sequencing. Metadata variables are depicted in grey and bacterial strains designated by colour. Chord links are coloured according to increased abundance (red) or decreased abundance (blue). The width of each chord is scaled according to the log2 fold changes in abundance. CCP, cyclic citrullinated protein; HLA, human leucocyte antigen; NORA, new-onset rheumatoid arthritis; RA, rheumatoid arthritis; RF, rheumatoid factor; V, versus.
Proportion of Prevotellaceae presence
Within the shotgun metagenomic data set, we assessed for the presence of the P. copri transposon described by Nii et al 41 but found no significant association with CCP positivity, disease progression or HLA status (online supplemental materials, page 50-51). However, we observed a higher number of transposon matches in individuals with specific Prevotella strains.
The shotgun sequencing data set comprising 27 CCP+ at risk (17 progressors), 7 NORA and 15 healthy controls corroborated an increase in Prevotellaceae species among at risk individuals relative to healthy controls (onlineonline supplemental materials, page 32-33) and strain-specific changes of Prevotellaceae associated with clinical variables (see online supplemental materials, page 35-39).
In CCP+ at risk individuals, gut microbiome alterations can begin 10 months before arthritis development
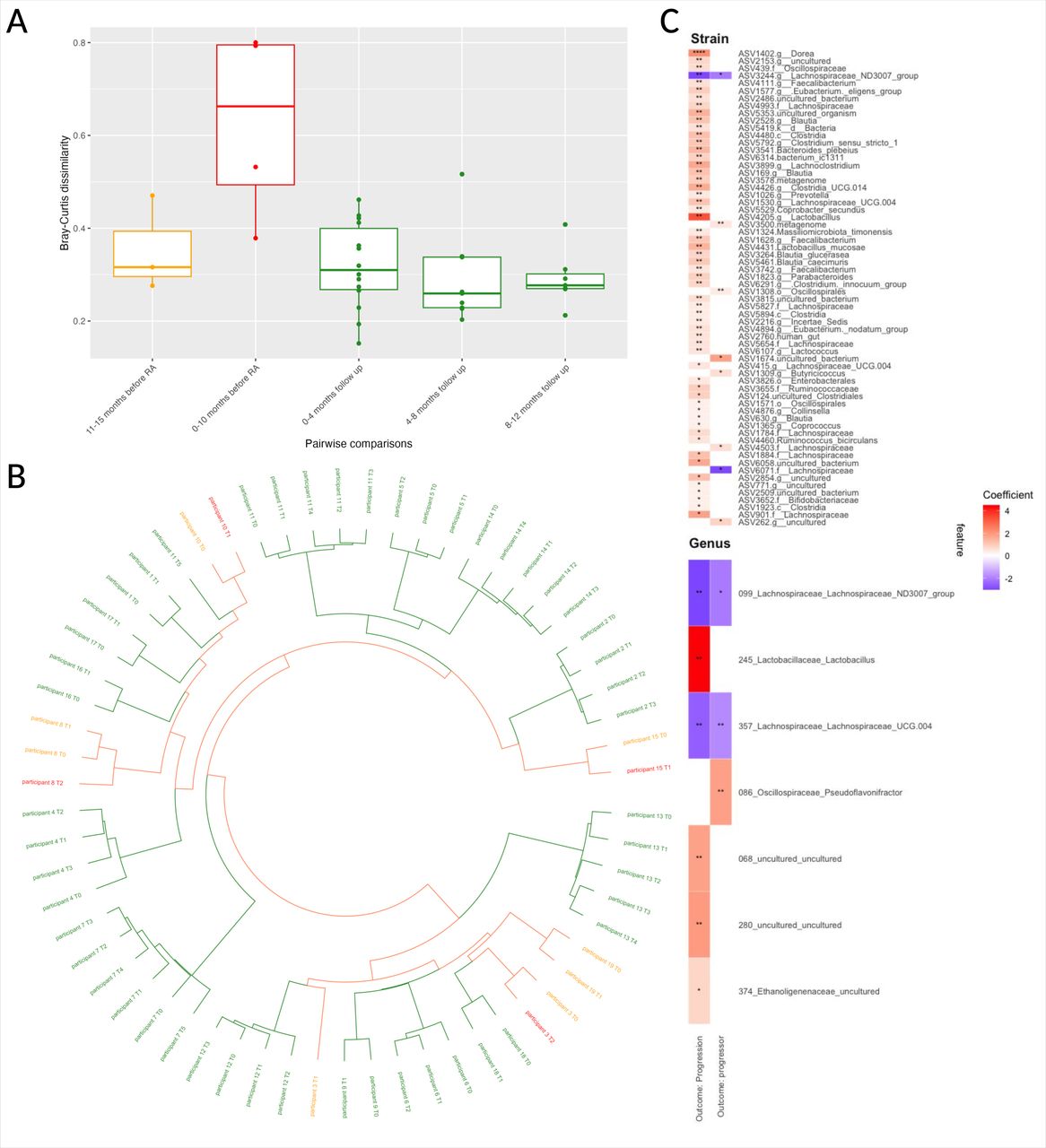
Due to the observed decrease in alpha diversity preceding RA onset (see online supplemental materials, page 44), pairwise dissimilarities between timepoints were assessed to examine gut microbiome stability using 16S rRNA amplicon sequencing. These dissimilarities were categorised into intervals related to arthritis development (ie, progression), either retrospective for progressors or prospective from baseline for non-progressors, and presented in figure 4A. Time to progression spanned 0–15 months preceding arthritis onset; the time for non-progressor from baseline to final follow-up was at least 12 months.

Temporal microbial analysis of CCP+ at risk individuals. (A) Pairwise Bray-Curtis dissimilarity boxplots from 16S rRNA amplicon sequencing. Boxplot of pairwise Bray-Curtis dissimilarities grouped according to time from progression: 0–10 months (n=4) and 11–15 months (n=3); or time from baseline for non-progressors: 0–4 months (n=14), 4–8 months (n=6) and 8–12 months (n=6) of follow-up. (B) Circulated dendrogram of longitudinal samples. Constructed using logged Bray-Curtis dissimilarity index and clustered using Ward D2 methodology. Samples coloured according to outcome: non-progressors (green), progressors (orange) and progression (red). (C) Mixed-effects linear modelling with bacterial profiles. Linear model of top 50 strains with significant associations in heatmap, plotted with coloured according to coefficient; p values are indicated as follows: *p<0.25, **p<0.05, ***p<0.01, ****p<0.001. Progression denotes sampling at diagnosis of RA. Progressor denotes sampling prior to progression. All plots constructed using 16S RNA amplicon sequencing. CCP, cyclic citrullinated protein; RA, rheumatoid arthritis.
CCP+ progressors (ie, those who developed arthritis) exhibited the most significant instability (p=0.044, BH-adjusted t-test), with median Bray-Curtis dissimilarities exceeding 0.6 in the period immediately leading up to arthritis onset. This pattern indicates that gut microbiome structural changes commence around 10 months before clinical arthritis development and may extend beyond diagnosis. In contrast, the period from 10 to 15 months prior to RA onset and among non-progressors demonstrated relative stability, with lower Bray-Curtis dissimilarities suggesting more consistent microbiome structures over time and less variability between samples. Clinical, serological and radiological findings from the longitudinal participants are provided in the online supplemental materials, page 40-43.
Hierarchical clustering demonstrates modest microbial divergence prior to RA onset
To determine if samples at the progression timepoint were structurally more similar to each other or to their respective preceding samples (16S rRNA amplicon sequencing), a hierarchical tree was constructed. The resulting circular dendrogram showed higher structural similarity within individual successive samples than between different individuals. However, a notable divergence was observed between progression samples and their immediate predecessors, indicated by increased branch lengths in the dendrogram, such as seen with participant 8 and participant 3 (figure 4B). This was further explored using a multidimensional scaling analysis (online supplemental materials, page 44-45), which corroborates dispersion of the coordinates prior to RA diagnosis.
The gut microbiome of individuals progressing to arthritis accumulates pathobionts over time
Mixed-effects linear models on 16S rRNA amplicon sequencing data were used to analyse temporal bacterial fluctuations in the gut microbiome as some individuals progressed to arthritis. Each participant was defined as the random effect to accommodate correlations across multiple timepoints. The models analysed data at both the genus and strain levels (figure 4C).
At the genus-level analysis, six taxa were associated with samples collected at the time of RA diagnosis, termed progression (figure 4C), while three taxa were associated to samples preceding RA onset, termed progressors. Significantly, two taxa from the Lachnospiraceae family showed consistent depletion in both progression and progressors samples (figure 4C). Strain-level analysis corroborated these findings, demonstrating a decrease in ASV3244, a specific Lachnospiraceae strain, in both progressors and at progression. In contrast, certain bacterial strains were found to increase in the gut microbiome of RA progressors, especially at the progression point (figure 4C). Detailed outputs are enumerated in the online supplemental materials, page 46-49. Notably, ASV1026, identified as Prevotella and enriched in the progression samples, was identified as P. copri through BLAST analysis.
NORA gut microbiome is associated with enhanced amino acid and energy metabolism
MaAsLin linear modelling (FDR q value <0.25) using shotgun metagenomic sequencing identified differential abundance in 18 MetaCyc pathways when comparing at risk individuals (n=27), individuals with NORA (n=7) and healthy controls (n=15), with healthy controls serving as the reference group for all comparisons. Eleven pathways exhibited elevation in the NORA gut microbiome, while six showed reduction in healthy controls, as presented in figure 5A. There were no differentially significant pathways identified in CCP+ at risk group when compared with NORA or healthy controls. The MaAsLin analysis also identified key microbial features, including MetaCyc pathways, enzymes and gene families (figure 5B), that differ between individuals in various CCP categories (very high, high, low) when compared with healthy controls (online supplemental materials, pages 50-51). Notably, some enzymes, such as cobalt precorrin-5B (C.1) methyltransferase, were significantly associated with more than one CCP category (see figure 5B). Functional analysis of HLA, RF and RA risk category did not yield significant results.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Functional analysis of enzymes, gene families and MetaCyc pathways from shotgun metagenomic sequencing. (A) Heatmap of MetaCyc pathways according to group. Heatmap represents MetaCyc pathways differentially abundant in NORA and healthy controls. (B) Heatmap of enzyme commission numbers, gene families and MetaCyc pathways of anti-CCP categories. Asterisks indicate the level of statistical significance: *p<0.25, **p<0.05, ***p<0.01. The colour gradient represents the coefficient values. Constructed using shotgun metagenomic data. CCP, cyclic citrullinated protein; NORA, new-onset rheumatoid arthritis.
Discussion
This study presents a detailed examination of the gut microbiome’s alterations before the onset of RA, revealing community-wide changes that extend beyond key organisms, both confirming and expanding on current literature. The work presented here represents, to the best of our knowledge, the largest, most homogenous RA at risk cohort, combining unique longitudinal and microbial variance analyses. Our study provides several novel insights, including decreased alpha diversity in the at risk phase, confirmation of Prevotella overabundance in at risk individuals and that Prevotella abundance appears to be associated with the underlying risk profile of the individual, which is consistent despite adjustment for common microbiome confounders. Finally, we propose a potential timeline for when gut microbiome changes occur as individuals develop RA.
Notably, we identified significantly decreased alpha diversity in CCP+ at risk individuals compared with healthy controls (figure 2A). This was previously unidentified in three similar studies.8 24 25 These differences between our previous work and this study could be explained by considerable different cohort size (25 vs 124 CCP+ at risk, respectively). Additionally, our initial study relied on older operational taxonomic unit (OTU) clustering, which is less sensitive than newer denoising methodologies. Both Alpizar-Rodriguez et al 8 and Gilbert et al 25 have previously investigated the at risk phase of RA but there are considerable differences between the RA at risk populations included in these studies. RA risk can be identified through a combination of factors, including antibody status, symptoms and genetics. The cohort examined by Alpizar-Rodriguez et al 8 was characterised by positivity for anti-CCP and/or RF, along with the presence of symptoms. These symptoms were either assessed through a disease screening questionnaire or identified via a diagnosis of undifferentiated arthritis (UA). This approach results in a more diverse at risk population, potentially more advanced in the RA disease continuum due to the inclusion of individuals with UA. Furthermore, it also relied on older OTU analysis. Gilbert et al 25 used the same population; however, risk was subdivided into four categories, with varying risks including genetics, autoantibody presence and/or symptoms, again representing a heterogeneous population.
Interestingly, we showed alpha diversity was stratified according to anti-CCP antibody level (figure 2A), where individuals with high and very high anti-CCP levels had significantly lower alpha diversity, as defined by both observed and Shannon diversity index. In contrast, Gilbert et al 25 did not identify reduced alpha diversity in those at risk of RA. However, within the symptomatic and asymptomatic autoimmune groups, 55% and 70%, respectively, were anti-CCP antibody-negative, reflecting a very different population from our study. While eligibility for the Leeds at risk cohort has been defined through anti-CCP antibody positivity, longitudinal follow-up has revealed a small number of individuals (n=11) transition from low titre positive (anti-CCP2 >2.99 IU/mL and <9 IU/mL) to a negative anti-CCP antibody status. Low titre individuals and those converting to negative titre show preserved alpha diversity, similar to healthy controls (figure 2A), which corresponds with the findings of Gilbert et al.25 Comparison of the NORA group with healthy controls did not show significantly decreased alpha diversity, and this may be reflective of the small numbers within this cohort and the heterogeneity in terms of antibody positivity (NORA 73% anti-CCP antibody-positive, 100% RF-positive). However, this result is in keeping with previous work, where others have failed to identify decreased alpha diversity, as measured by Shannon diversity index in NORA compared with healthy controls.9 11 It is worth noting that we also observed decreased Shannon diversity with steroid use. However, given steroid use is pronounced in the symptomatic group (which tends to have higher CCP titres, HLA and RF positivity), it is impossible to know what portion of change is disease-related versus drug-related.
To understand the factors influencing microbial variability, a permutational MANOVA was conducted. This analysis identified age, vegetable intake and sequencing run as potential confounders (refer to figure 2B). Age stratification, ranging from 22 to 80 years in this cohort, aligns with existing literature42–44 as a critical factor in microbiome–disease correlations and was therefore incorporated into the statistical model. However, vegetable intake, while identified as a potential confounder, was not adjusted for in the model to avoid overcorrection that may obscure significant microbiome–disease associations. Considering the hypothesis that diet, including vegetable consumption, might influence the gut microbiome’s role in RA-related autoimmunity, adjusting for vegetable intake could potentially conceal key bacterial associations that are either protective or harmful. Consequently, the study refrained from adjusting for vegetable intake in the analyses. The same rationale was applied to adjustment of intestinal barrier integrity (zonulin family peptide; see figure 2B). Given the relatively high frequencies of non-steroidal anti-inflammatory drugs, proton pump inhibitors and steroid use within the at risk phases of RA, we investigated their effect on the gut microbiome. None of these variables showed a significant effect in the permutational multivariate analysis, likely due to the pro re nata use of medications coded as ‘ever used’ rather than ‘current use’.
The study highlights a distinct pattern of strain-specific enrichment and concurrent depletion of Prevotellaceae in the RA at risk phase, aligning with previous findings of Prevotellaceae overabundance in CCP+ at risk individuals compared with healthy controls (figure 2C). A specific strain of Prevotellaceae, ASV2058, showed significant enrichment in the CCP+ at risk group compared with healthy controls. BLAST analysis suggested this strain is likely P. copri. Notably, this enrichment was also observed in individuals with NORA compared with healthy controls. Intriguingly, ASV2058 was found to be less abundant in progressors versus non-progressors. Its abundance increased with higher CCP antibody titres and HLA positivity, but decreased in individuals with abnormal T-naïve cells. ASV1867, a second putative P. copri strain, was increased in progressors at baseline, positively correlated with CCP titres and abnormal T cells, but was negatively associated with HLA SE positivity. These findings might suggest that different strains of P. copri may have diverse roles in RA progression. One interpretation of these findings could be that ASV2058 is an additional risk factor for RA, associated with known serological and genetic risk markers, and in this context a lower threshold is required to induce early immune dysregulation and progression. Conversely, ASV1867 is not associated with genetic risk, and in this context relatively higher amounts are required for early immune dysregulation and RA progression.
Longitudinal analysis revealed that RA progressors tend to have a more unstable gut microbiome (figure 4A) compared with non-progressors. A median Bray-Curtis dissimilarity exceeding 0.6 was observed in progressors within 10 months prior to RA diagnosis (figure 4A), indicating a considerable shift in the gut microbiome during this period. When comparing the time periods of 0–10 months before RA and 11–15 months before RA, the gut microbiome appears relatively stable until 10 months before RA onset (figure 4A). The median Bray-Curtis index for the period of 11–15 months (0.3) aligns with values observed in non-progressors, suggesting significant changes may occur closer to RA onset. This increase in Bray-Curtis values during the period of 0–10 months may reflect the influence of RA development and associated inflammatory responses on the gut microbiome. However, for participant 3, both samples taken within 9 months before RA diagnosis showed a Bray-Curtis dissimilarity index of 0.8 (figure 4A), despite clinical reviews excluding RA progression at 9 and 6 months prior. This indicates that changes in the gut microbiome were occurring before the onset of clinical arthritis, although environmental influences cannot be ruled out. Similar patterns of beta diversity volatility have been noted in patients with inflammatory bowel disease around flare-ups, where increased instability correlated with heightened inflammation.45
Our finding of increased putrescine degradation and threonine synthesis in the NORA cohort (figure 5A) is consistent with previous work.9 11 Pathways such as L-lysine degradation, L-threonine metabolism and the tricarboxylic acid (TCA) cycle (acetate producers) were notably more active in the NORA group compared with healthy controls, suggesting an upregulation of amino acid metabolism and energy production in these individuals. Additionally, pathways involved in glycol metabolism and degradation, as well as phenylacetate degradation, were also elevated in individuals with NORA, indicating potential alterations in energy utilisation and aromatic compound processing. These findings suggest that individuals with NORA exhibit distinct metabolic shifts, particularly in pathways related to amino acid and energy metabolism, which could reflect underlying differences in metabolic demands or stress responses associated with inflammation.
Investigation of gut microbiome function in relation to anti-CCP antibody titre identified associations between several enzymes, gene families and metabolic pathways, aligning with literature showing that ACPA antibody titre affects microbiome function.46 For instance, the increase of type IV secretory system conjugative DNA transfer family protein, XRE family transcriptional regulator, DNA topoisomerase and excisionase in CCP+ high and low individuals suggests a coordinated alteration in microbial gene expression. Additionally, enzymes such as cobalt precorrin-5B (C.1) methyltransferase, also elevated in both CCP+ high and low individuals, responsible for the biosynthesis in cobalamin (vitamin B12), indicate potential adjustment in microbial vitamin synthesis. The activity of other enzymes, such as cadmium-exporting ATPase and pyridoxal kinase, points to altered metal ion homeostasis and coenzyme metabolism. Histidine degradation, inosine 5'-phosphate biosynthesis and folate transformations reveal alterations in amino acid metabolism, nucleotide synthesis and one carbon metabolism (oxidative stress), reflective of our findings in the NORA group. It is worth noting, although comparable dietary quality scores suggest that there were no major deviations in diet quality, including protein intake. We do recognise that direct quantification of protein consumption could provide more precise insights into its potential impact on amino acid metabolism.
We specifically assessed for the presence of the transposon described by Nii et al 41 in our shotgun metagenomics data. Our results did not show any significant relationship between the presence of this transposon and clinical factors such as CCP positivity, disease progression or HLA status (online supplemental materials, page 51-52). The lack of association with these variables suggests that while Prevotella strains with this transposon are indeed present in the cohort (including healthy controls) and potentially relevant, their presence alone does not correlate with these specific clinical outcomes in our cohort. We did identify a higher number of transposon matches in individuals who possessed our identified Prevotella strains of interest. This suggests an interesting association between the presence of specific Prevotellaceae strains which are associated with RA clinical variables and progression and an increased number of matches to a P. copri transposon sequence, and raises the question of whether this is merely a reflection of overall Prevotella abundance or if it points to a more specific biological relationship; this warrants further work beyond the scope of this paper.
The limitations of this research, particularly the small longitudinal sample size and the lack of 1:1 longitudinal comparison between CCP+ at risk and healthy controls, temper the generalisability of the results and increase the chance of spurious associations. This should be considered when interpreting the findings and calls for further research with larger longitudinal cohorts. The heterogeneity in the NORA cohort reflects the practical constraints of recruitment from standard of care clinics, which may limit the interpretability of direct comparisons. Moreover, extending the analysis beyond the bacterial microbiome to include viromes, mycobiomes and archaeomes could unveil additional facets of microbial influence on RA pathogenesis. The absence of integrated transcriptomic or metabolomic data restricts our interpretation to potential rather than confirmed metabolic activity.
In summary, we have shown that individuals at risk of RA harbour a distinctive gut microbial composition, including but not limited to an overabundance of Prevotellaceae species. This microbial signature is consistent and correlates with traditional RA risk factors. Longitudinal examination shows a dynamic microbial environment preceding RA onset. Further research into this late phase of disease development is merited, especially given the potential of the gut microbiome as a target for prevention, including in high-risk individuals with imminent arthritis.
Data availability statement
Raw sequencing data available upon reasonable request from the corresponding author.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was conducted at Chapel Allerton Hospital (CAH), Leeds, UK, with ethical approval granted by the Leeds, West Yorkshire Research Ethics Committee (reference: 06/Q1205/169). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We extend our gratitude to the participants of the Leeds at risk cohort and the patient discussion groups. We also wish to acknowledge the work of Professor F Ponchel with T cell subset analysis and Professor A Kastbom with ZFP quantification. Figure 1 was created with BioRender.com.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Josef S Smolen
X @DrCRooney
Contributors The study was designed by PE, KM, MHW and CMR. Sample and data collection was conducted by KM and CMR. Sample processing was completed by CMR. Data analysis was completed by CMR and supervised by IBJ. All authors contributed to writing the manuscript and approved it for publication. CMR is the guarantor.
Funding CMR was funded to complete this work by a personal fellowship from Versus Arthritis (22294) and Leeds Cares (9R01-A1210) and is currently funded by a National Institute for Health Research (NIHR) Clinical Lectureship. The Leeds CCP clinic is supported by the NIHR Leeds Biomedical Research Unit.
Competing interests CMR has received funding from Versus Arthritis and Leeds Cares and in-kind support from 4D Pharma PLC. KM has received grants from Gilead, Lilly, Serac Healthcare, AstraZeneca and DeepCure; consulting fees from AbbVie, UCB, Lilly, Galapagos, Serac Healthcare, Zura Bio and DeepCure; and honoraria from AbbVie, UCB, Lilly, Galapagos, Serac Healthcare, Zura Bio and DeepCure. PE has received grants from AbbVie, BMS, Lilly, Novartis, Pfizer and Samsung; and consulting fees from AbbVie, Activia, AstraZeneca, BMS, Boehringer Ingelheim, Galapagos, Gilead, Immunovant, Janssen, Lilly and Novartis.
Patient and public involvement Before starting this research, patients contributed to the study's design and feasibility, influencing recruitment, sample return and dissemination of outputs through a patient-focused discussion group at Chapel Allerton Hospital, Leeds. During the study, participant feedback on the stool collection kit was gathered and led to its redesign.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.